¶ Nuclear Magnetic Resonance (NMR)
Einsatz von NMR zur berührungslosen Bestimmung von Nährstoffen in landwirtschaftlichen Substraten.
¶ Beschreibung
NMR (nuclear magnetic resonance) - zu Deutsch, Kernspinresonanz - beschreibt den Effekt, dass sich das magnetische Moment eines Atomkerns in einem äußeren (künstlichem) Magnetfeld ausrichtet und bei Störung bzw. Anregung mit einer bestimmten Eigenfrequenz präzessiert (taumelt). Damit verbunden ist eine Ausstrahlung oder Aufnahme von Radiofrequenzwellen, welche mit den Kerneigenschaften und damit Elementart und der Magnetfeldstärke verbunden ist.
Für die Anwendung in der Landwirtschaft wird dies genutzt, um Elemente, wie z. B. Stickstoff (N) in ihren Verbindungen zu bestimmen und quantitativ nachzuweisen.
¶ Abgrenzung zu anderen NMR-Anwendungen
Im Allgemeinen wird NMR für die Strukturauflösung von Molekülen, Nachweis von Substanzen und als bildgebendes Verfahren (MRT – Magnet Resonanz Tomografie) eingesetzt. Hierbei werden die Einflüsse durch wechselnde äußere Magnetfelder ausgenutzt, entweder durch die sogenannte chemische Umgebung – also die Anordnung der Atome im Molekül – oder gezielte örtliche und zeitliche Änderung des künstlich erzeugten Magnetfeldes.
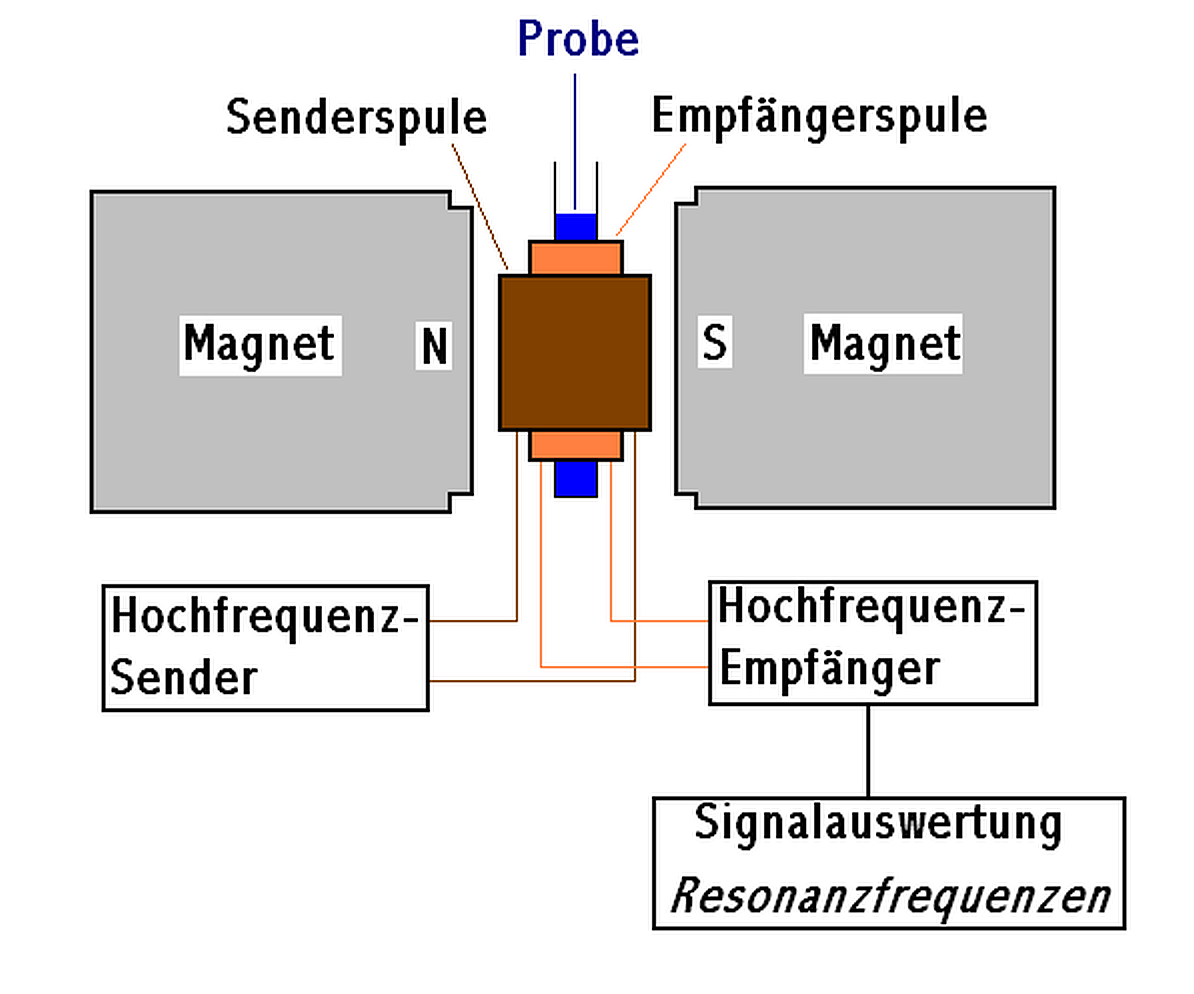
Für die landwirtschaftliche Anwendung zur Konzentrationsbestimmung haben diese Methoden jedoch keine Bedeutung: Es geht lediglich um die Messung der Signalstärke, hervorgerufenen durch NMR aktive Verbindungen, in einem konstanten Magnetfeld. Während stationäre NMR Geräte das Magnetwechselfeld durch supraleitende Elektromagnete erzeugen, dient in mobilen Laborgeräten ein Permanentmagnet zur Erzeugung eines konstanten Feldes von etwa 1,5 Tesla [2].
¶ Signalentstehung
Damit ein Atomkern NMR aktiv ist, muss er einen Kernspin ungleich Null haben, da dieser das magnetische Moment erzeugt. Dies ist bei Atomkernen mit ungerader Protonen- und/oder Neutronenanzahl der Fall, wie 14N, 17O, 31P und 39K.
Werden nun die magnetischen Momente der Atomkerne einem äußeren Magnetfeld ausgesetzt, so richten sie sich wie Kompassnadel daran aus. Dieser Zustand ist mit einer spezifischen Energie verknüpft, die aber wegen der Quanteneigenschaften nur bestimmte Niveaus annehmen kann. Beispielsweise entweder parallel oder anti-parallel zu den Feldlinien des äußeren Magnetfeldes für Kerne mit Spin 1/2. Dies entspricht zwei unterschiedlichen Energiezuständen des Atoms, deren Übergang durch elektromagnetische Strahlung verbunden ist mit Frequenzen im Radiowellenbereich von einigen MHz.
Bei dem Messvorgang einer Probe, welche sich im Messraum eines NMR-Spektrometers in einem (möglichst homogenen) Magnetfeld befindet, geschieht nun Folgendes:
Vor der eigentlichen Messung wird durch einen oder auch mehrere (da das Magnetfeld vom Puls sehr klein gegenüber dem statischen, äußeren Magnetfeld ist, reicht ein Puls nicht aus) kurzen Radiowellen-Puls von typischerweise einigen µs-Länge mit der Resonanzfrequenz das magnetische Moment der Atomkerne in der Probe relativ zum äußeren Magnetfeld gekippt – am besten um 90° und präzedieren danach mit der Resonanzfrequenz, um die Richtung des äußeren Magnetfeldes. Die Atomkerne orientieren sich nach einer gewissen Relaxationszeit (typischerweise einige ms) unter Emission von einer Radiowellen-Strahlung mit eben dieser Resonanzfrequenz und spezifischer Intensität wieder am äußeren Magnetfeld [2].
Diese Emission wird als eigentliches Messsignal detektiert. Ihre Signalstärke ist im Allgemeinen sehr schwach. Damit ein nutzbares Signal-zu-Rausch-Verhältnis entsteht, wird der Prozess der Anregung und Messung bis zu einige Stunden lang wiederholt. Um verschiedene Elementarten zu detektieren, müssen verschiedene Resonanzfrequenzen gewählt und auch diese zeitlich nacheinander durchgeführt werden, womit sich die Messzeit wiederum verlängert [3].
Die Signalstärke ist nicht nur von der Element-Konzentration abhängig, sondern auch von der Stärke des Magnetfeldes. Zwar beeinflusst die Magnetfeldstärke in erster Linie direkt proportional die Differenz des Energieniveaus und damit die Resonanzfrequenz; wenn diese aber sehr gering gegenüber der thermischen Energie bei Raumtemperatur ist, führt das zu einer nahezu Gleichbesetzung der beteiligten Energieniveaus (Boltzmann-Verteilung). Somit stehen nur sehr wenige Atomkerne zur Anregung vom unteren Niveau ins Höhere zur Verfügung. Eine höhere Magnetfeldstärke und damit höhere Resonanzfrequenz bewirkt eine höhere Verteilung der Atomkerne im unteren Energieniveau, sodass mehr Atomkerne aus dem oberen Niveau wieder zurück relaxieren können und dabei ein stärkeres Signal ausstrahlen.
¶ Apparatives und Messdauer
In den folgenden Abbildungen (Abbildung 1 & 2) ist das Gerät der Firma NanoNord abgebildet, welches eine Magnetfeldstärke von 1,6 Tesla besitzt. Die Probe wird in den zylinderförmigen Messraum von ca. 45 mm Länge und 10 mm Durchmesser mit einer Plastikröhre (Abbildung 3) eingeführt. Dies kann durch einen Probentauscher (Sample Changer) automatisiert werden.


Die typischen Messzeiten richten sich nach dem Signal-zu-Rausch-Verhältnis bei dem gewünschten Inhaltsstoff und Probenart. Eine Vervierfachung der Messzeit führt zu einer Verdoppelung der Genauigkeit (Halbierung der Mess-Standardabweichung)[3]. Bei Flüssigmist liegt er für die Stickstoffverbindungen bei etwa 30 min, für Phosphor bei 60 min, während das Sauerstoffisotop 17O zwar einen geringen Anteil gegenüber dem 16O im natürlichen Vorkommen hat, dafür aber in einer wässrigen Probe als Bestandteil von Wasser H₂O ein starkes Signal liefert. Dies kann z. B. für die Bestimmung der Trockensubstanz benutzt werden.
Allgemein ist die Signalstärke von der Konzentration des betreffenden Elementes in der Probe, seiner Isotopenverteilung und der „Stärke“ des Energieniveauüberganges, welches typischerweise als Verhältnis zu dem Wasserstoffsignal angegeben wird, abhängig. Zudem ist die Signalstärke noch über die Boltzmann-Verteilung der beteiligten Energieniveaus bei gegebener Temperatur auch von der Resonanzfrequenz und damit wieder vom äußeren Magnetfeld abhängig.
Eine Besonderheit dieser Messtechnik ist noch, dass nicht alle Atome zum Signal beitragen, sondern z. B. bei Stickstoff (N) die festgebundene Form als Norg nicht zum Signal beiträgt und über die Resonanzfrequenz von 14N nur NH4+ gemessen wird. Der organische Stickstoffanteil (Norg) kann über die Bestimmung der Trockensubstanz (TS) durch 17O, aufgrund der engen Korrelation zwischen Norg und TS, errechnet werden. Während bei Phosphor die Signalstärke groß genug ist, um den gelösten Anteil P04- als auch den gebundenen Anteil zu bestimmen. Diese Phosphorfraktionen lassen sich durch unterschiedliche Relaxationszeiten differenzieren [2].
Die Messung der unterschiedlichen Elemente erfolgt sequentiell nacheinander, da sie unterschiedliche Resonanzfrequenzen haben. Dieser Effekt summiert die einzelnen Messzeiten, sodass für die Bestimmung von Ammonium (NH4+), Gesamtstickstoff (Ntot), Phosphat (PO4-), Gesamtphosphor (Ptot), pH und TS je nach Genauigkeitsanspruch mehrere Stunden benötigt werden.
¶ Anwendungsbereich
Hier folgen passende Praxisbeispiele von FARMPRAXIS.
¶ Quellen
- NanoNord
- NPK NMR Sensor: Online Monitoring of Nitrogen, Phosphorus, and Potassium in Animal Slurry: M. Sörensen, O. Jensen, O. N. Bakharev, T. Nyord, C. Nielsen
- Fast and Accurate Quantification of Nitrogen and Phosphorus Constituents in Animal Slurries Using NMR Sensor Technology: O. N. Jensen, M. Beyer, M. K. Sörensen, M. Kreimeyer, N.C. Nielsen
- NMR-Spektrometer: Oguenther at Wikipedia
¶ Autoren
Eiko Thiessen, Christian-Albrechts-Universität Kiel, Institut für Landwirtschaftliche Verfahrenstechnik, Max-Eyth-Str. 6, 24118 Kiel